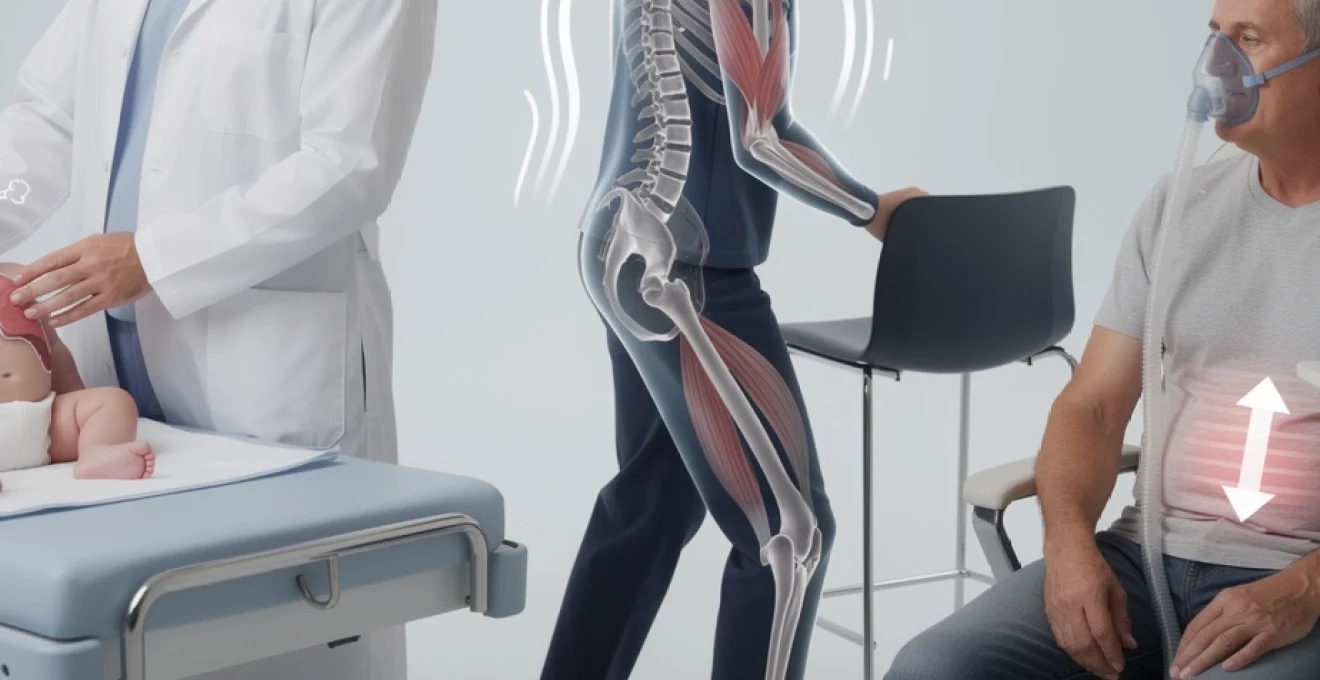
La maladie de Pompe représente une pathologie génétique rare qui touche environ 5 000 à 10 000 personnes dans le monde, avec près de 200 cas diagnostiqués en France selon le registre national. Cette glycogénose de type II résulte d’un déficit enzymatique qui provoque une accumulation progressive de glycogène dans les cellules musculaires, entraînant des conséquences potentiellement graves sur la fonction cardiaque, respiratoire et motrice. Reconnaître précocement les signes cliniques caractéristiques constitue un enjeu majeur pour vous permettre d’orienter rapidement vers une prise en charge spécialisée. Les manifestations varient considérablement selon l’âge d’apparition, allant de l’hypotonie sévère chez le nourrisson à une faiblesse musculaire progressive chez l’adulte. Comprendre ces symptômes d’alerte peut littéralement sauver des vies ou préserver durablement votre qualité de vie.
Physiopathologie de la glycogénose de type II : comprendre le déficit enzymatique en alpha-glucosidase acide
La maladie de Pompe trouve son origine dans des mutations du gène GAA localisé sur le chromosome 17q25.3, qui code pour l’alpha-glucosidase acide, également appelée maltase acide. Cette enzyme lysosomale joue un rôle crucial dans le métabolisme énergétique cellulaire en dégradant le glycogène en glucose au sein des lysosomes. Ces organites fonctionnent comme de véritables centres de recyclage cellulaire, similaires à des usines de transformation qui traitent continuellement les déchets métaboliques pour maintenir l’homéostasie cellulaire.
Lorsque l’activité de l’alpha-glucosidase acide est réduite ou absente, le glycogène s’accumule progressivement dans les lysosomes, provoquant leur engorgement puis leur dysfonctionnement. Cette surcharge lysosomale affecte particulièrement les cellules musculaires, qui possèdent des besoins énergétiques importants et un métabolisme glycogénique actif. L’accumulation pathologique de glycogène perturbe l’architecture cellulaire normale, compromet la fonction contractile et déclenche des processus d’autophagie dysfonctionnelle qui aggravent encore la détérioration musculaire.
Plus de 150 mutations différentes du gène GAA ont été identifiées à ce jour, expliquant la grande hétérogénéité clinique observée. Dans la forme infantile classique, l’activité enzymatique résiduelle est quasi nulle, inférieure à 1% de la normale, tandis que dans les formes tardives, elle peut atteindre 10 à 30% de l’activité normale. Cette corrélation entre le degré de déficit enzymatique et la sévérité clinique n’est toutefois pas absolue, certains facteurs génétiques modificateurs et environnementaux influençant également la progression de la maladie. Comprendre ces mécanismes pathologiques vous permet d’anticiper les complications potentielles et d’adapter la surveillance clinique en fonction du profil enzymatique.
Manifestations cliniques de la forme infantile classique : hypotonie néonatale et cardiomyopathie hypertrophique
La forme infantile de la maladie de Pompe se révèle généralement entre l’âge de un et six mois, bien que le nouveau-né apparaisse normal à la naissance. Cette période de latence correspond au temps nécessaire pour que l’accumulation glycogénique atteigne un seuil critique perturbant la fonction cellulaire. Les premiers signes cliniques évocateurs doivent immédiatement vous alerter car le pronostic dépend directement de la précocité du
prise en charge. Ils associent le plus souvent une hypotonie marquée, des difficultés d’alimentation, une cardiomégalie et des troubles respiratoires. Sans traitement spécifique précoce par enzymothérapie substitutive, l’évolution vers l’insuffisance cardiaque et respiratoire est rapide, avec un risque vital majeur avant l’âge de 2 ans.
Syndrome du bébé mou : faiblesse musculaire axiale et retard des acquisitions motrices
Le signe clinique le plus frappant de la forme infantile classique est le syndrome du bébé mou. Le nourrisson présente une hypotonie axiale importante : lorsqu’on le prend dans les bras, il « glisse », a du mal à tenir sa tête et adopte une posture dite en « grenouille », avec les cuisses écartées et les hanches fléchies. Cette faiblesse musculaire concerne surtout les muscles du tronc et des ceintures, ce qui explique le retard des acquisitions motrices.
Concrètement, vous pouvez observer que le bébé peine à se retourner, à se redresser en position assise ou à maintenir la tête dans l’axe. À l’âge où un enfant sain commence à se retourner ou à s’asseoir avec appui, l’enfant atteint de maladie de Pompe reste très passif et peu tonique. Ce retard moteur n’est pas isolé : il s’accompagne souvent d’une fatigabilité rapide, de pleurs faibles et d’une difficulté à bouger activement les membres contre la pesanteur.
Sur le plan clinique, les réflexes ostéo-tendineux peuvent être diminués, mais la sensibilité reste normale, ce qui aide à distinguer la maladie de Pompe d’une atteinte neurologique centrale. Face à un nourrisson très hypotonique, avec un tableau de « floppy baby » et une cardiomégalie associée, la glycogénose de type II doit systématiquement faire partie des diagnostics évoqués et conduire à un dosage rapide de l’activité enzymatique GAA.
Atteinte cardiaque précoce : hypertrophie ventriculaire gauche et insuffisance cardiaque congestive
La cardiomyopathie hypertrophique est un marqueur majeur de la forme infantile classique de la maladie de Pompe. L’accumulation massive de glycogène dans les cardiomyocytes entraîne une hypertrophie concentrique du ventricule gauche, visible à l’échocardiographie et souvent déjà suspectée sur la radiographie thoracique par une cardiomégalie importante. Cette hypertrophie s’accompagne d’un épaississement des parois et d’une altération progressive de la fonction systolique et diastolique.
Cliniquement, vous pouvez observer des signes d’insuffisance cardiaque congestive : tachycardie, difficultés à s’alimenter, sueurs abondantes au moindre effort, prise de poids insuffisante malgré une alimentation adaptée. Des râles crépitants, une hépatomégalie de stase et des troubles du rythme cardiaque peuvent également apparaître. Dans certains cas, la maladie se révèle brutalement par une décompensation cardiaque aiguë ou une mort subite liée à des troubles de conduction.
L’électrocardiogramme montre classiquement un raccourcissement de l’intervalle PR, une hypertrophie ventriculaire gauche et parfois des troubles de repolarisation. L’échocardiographie confirme l’hypertrophie ventriculaire diffuse, parfois associée à une obstruction dynamique du tractus d’éjection. Comprendre ce profil cardiologique typique permet de distinguer la maladie de Pompe d’autres cardiomyopathies métaboliques ou structurelles et de mettre en route sans délai l’enzymothérapie substitutive, qui peut réduire significativement l’épaisseur myocardique.
Hépatomégalie massive et macroglossie : signes d’accumulation glycogénique viscérale
Au-delà des muscles squelettiques et cardiaques, la maladie de Pompe infantile touche aussi plusieurs organes viscéraux, en particulier le foie et la langue. L’hépatomégalie est fréquente : le foie est augmenté de volume, ferme, et peut être palpé plusieurs centimètres sous le rebord costal. Cette augmentation de taille est liée à la surcharge en glycogène des hépatocytes et des cellules de Kupffer. Elle n’est généralement pas associée à une insuffisance hépatique sévère, mais peut s’accompagner d’une légère élévation des transaminases.
La macroglossie (langue volumineuse) est un autre signe d’appel important. Elle se manifeste par une langue proéminente, parfois en permanence entre les lèvres, qui gêne la succion, la déglutition et plus tard l’articulation des sons. Pour vous, parents ou soignants, une langue nettement trop grosse, associée à une hypotonie globale et à une cardiomégalie, doit faire suspecter une maladie de surcharge, dont la glycogénose de type II.
Ces signes viscéraux d’accumulation glycogénique ne sont pas spécifiques à la maladie de Pompe, mais leur association à une myopathie et à des troubles cardiaques crée une constellation clinique très évocatrice. Ils justifient la réalisation rapide d’examens biologiques ciblés, notamment un dosage enzymatique GAA et un bilan hépatique, afin de ne pas retarder le diagnostic et la prise en charge.
Détresse respiratoire par faiblesse des muscles intercostaux et du diaphragme
La fonction respiratoire est rapidement compromise dans la forme infantile de la maladie de Pompe, principalement en raison de la faiblesse des muscles intercostaux et du diaphragme. L’enfant respire vite, avec un tirage costal, des battements des ailes du nez et parfois un geignement expiratoire. Les infections respiratoires sont fréquentes et graves car le nourrisson a du mal à tousser efficacement, à évacuer les sécrétions et à ventiler correctement les bases pulmonaires.
Vous pouvez remarquer un essoufflement au moindre effort, un refus de téter lié à la dyspnée, ou encore une cyanose des lèvres lors des pleurs prolongés. La position allongée peut majorer la gêne respiratoire, poussant l’enfant à adopter spontanément une posture légèrement surélevée. Sans assistance ventilatoire adaptée, cette faiblesse des muscles respiratoires progresse vers une insuffisance respiratoire chronique, cause fréquente d’hospitalisations répétées et de décompensations aiguës.
Les gaz du sang peuvent montrer une hypercapnie et une hypoxémie, tandis que la radiographie thoracique révèle parfois un diaphragme bas et peu mobile. Dès les premiers signes de détresse respiratoire, une prise en charge spécialisée en pneumologie pédiatrique s’impose, associant kinésithérapie respiratoire, surveillance des gaz du sang et, si besoin, ventilation non invasive. Là encore, plus le traitement spécifique de la maladie de Pompe est débuté tôt, plus la dégradation de la fonction respiratoire peut être ralentie.
Symptômes de la maladie de pompe à début tardif chez l’enfant et l’adulte
À l’inverse de la forme infantile, la maladie de Pompe à début tardif (enfant, adolescent ou adulte) évolue de manière plus lente et hétérogène. Vous pouvez parfois vivre plusieurs années avec des symptômes discrets avant que le diagnostic ne soit posé. Pourtant, derrière une simple « fatigue musculaire » ou des difficultés à monter les escaliers peut se cacher une glycogénose de type II évolutive, avec un risque d’insuffisance respiratoire à long terme.
Les principaux signes d’alerte sont une faiblesse des muscles proximaux (épaules, hanches), une incapacité progressive à courir, sauter ou se relever du sol, mais aussi des troubles respiratoires nocturnes insidieux (maux de tête matinaux, somnolence diurne, ronflements, apnées). Selon l’âge d’apparition, on distingue une forme juvénile (avant 20 ans) et une forme de l’adulte (souvent entre 30 et 50 ans), mais le spectre clinique est continu. Plus les symptômes débutent tôt, plus la maladie a tendance à progresser rapidement.
Myopathie proximale progressive : faiblesse des ceintures scapulaire et pelvienne
La myopathie proximale est la manifestation cardinale de la maladie de Pompe tardive. Elle touche d’abord les muscles des ceintures pelvienne et scapulaire : vous pouvez avoir du mal à vous relever d’une chaise sans vous aider des bras, à monter les escaliers, à porter des charges au-dessus de la tête ou à vous pencher pour ramasser un objet au sol. Chez l’enfant, on observe une perte progressive de compétences motrices acquises, comme courir ou sauter, avec un recours plus fréquent aux mains pour se relever (signe de Gowers).
La marche devient progressivement dandinante, avec un balancement des hanches (démarche de Trendelenburg) lié à la faiblesse des muscles fessiers. Les chutes et faux-pas se multiplient, surtout sur terrain irrégulier ou lors des changements de direction rapides. Des douleurs musculaires diffuses, en particulier au niveau des cuisses et du bas du dos, peuvent accompagner la faiblesse, mais elles ne sont pas toujours au premier plan.
Ce tableau de myopathie proximale progressive peut mimer d’autres pathologies, comme une dystrophie musculaire des ceintures ou une myopathie inflammatoire. C’est pourquoi il est essentiel de signaler aussi tout symptôme respiratoire associé, même discret, et de demander explicitement à votre médecin si une maladie de Pompe a été envisagée. Un simple dosage enzymatique orienté peut alors changer la trajectoire de la prise en charge.
Insuffisance respiratoire restrictive nocturne : hypoventilation alvéolaire et apnées du sommeil
Dans la forme tardive de la maladie de Pompe, l’atteinte respiratoire est parfois plus invalidante que la faiblesse motrice elle-même. La faiblesse des muscles respiratoires entraîne une insuffisance respiratoire restrictive, qui se manifeste d’abord la nuit. Pourquoi la nuit ? Parce qu’en décubitus dorsal, le diaphragme doit travailler davantage contre la pesanteur, alors même que son efficacité est altérée.
Vous pouvez ressentir des maux de tête au réveil, une somnolence diurne, une fatigue inexpliquée malgré des nuits apparemment complètes, des réveils nocturnes avec sensation d’étouffement ou de palpitations. Votre entourage peut remarquer des ronflements intenses, des pauses respiratoires (apnées du sommeil) ou une respiration superficielle. Ces signes traduisent une hypoventilation alvéolaire nocturne, avec une accumulation de dioxyde de carbone dans le sang.
Sans prise en charge, cette hypoventilation peut s’aggraver et se prolonger en journée, aboutissant à une insuffisance respiratoire chronique. Un bilan fonctionnel respiratoire (EFR), associé à une polysomnographie, permet de mettre en évidence une réduction de la capacité vitale et des épisodes d’apnées ou d’hypopnées. La mise en place d’une ventilation non invasive nocturne (type PPC ou BiPAP) améliore souvent de façon spectaculaire la qualité de vie, en attendant ou en parallèle de l’enzymothérapie substitutive.
Atteinte paravertébrale : lordose lombaire excessive et syndrome de beevor
La maladie de Pompe à début tardif touche aussi les muscles paravertébraux, responsables du maintien de la posture. Leur faiblesse entraîne une hyperlordose lombaire (cambrure exagérée du bas du dos) ou parfois une scoliose, surtout chez les enfants et les adolescents en croissance. Vous pouvez ressentir des douleurs dorsales chroniques, une difficulté à rester debout longtemps et une sensation de « fatigue du dos » en fin de journée.
Un signe clinique intéressant, bien que méconnu, est le syndrome de Beevor. Lorsque le patient essaie de relever la tête ou le tronc en décubitus dorsal, l’ombilic se déplace vers le haut en raison de la faiblesse des muscles abdominaux inférieurs. Ce signe traduit une atteinte dissymétrique des muscles de la paroi abdominale et peut être particulièrement évocateur de myopathies affectant les muscles paravertébraux et abdominaux, dont la maladie de Pompe.
Sur l’IRM musculaire, on observe souvent une infiltration graisseuse prédominante des muscles spinaux lombaires, alors que certaines masses musculaires périphériques sont relativement préservées. Cette atteinte paravertébrale spécifique peut aider à distinguer la maladie de Pompe d’autres dystrophies musculaires. Pour vous, patients et proches, retenir que « un dos très cambré, douloureux et fatigable » peut être un signe d’alerte est déjà un premier pas vers un diagnostic plus rapide.
Dysfonction diaphragmatique : orthopnée et paradoxe respiratoire abdominal
Le diaphragme est le principal muscle de la respiration. Dans la forme tardive de la maladie de Pompe, il est souvent atteint de manière précoce, parfois avant même que la faiblesse des membres ne soit évidente. Comment suspecter une dysfonction diaphragmatique au quotidien ? En prêtant attention à certains signes simples : gêne respiratoire en position allongée (orthopnée), besoin de dormir avec plusieurs oreillers, essoufflement disproportionné pour de petits efforts.
Lors de l’examen clinique, le médecin peut observer un paradoxe respiratoire abdominal : à l’inspiration, l’abdomen se creuse au lieu de se gonfler, signe que le diaphragme ne se contracte plus efficacement et que la cage thoracique se soulève de manière compensatoire. Ce phénomène est particulièrement visible en position couchée. Des examens complémentaires, comme la mesure de la pression inspiratoire maximale ou une échographie diaphragmatique, peuvent confirmer l’atteinte.
Prendre en compte précocement la dysfonction diaphragmatique permet d’anticiper les complications respiratoires, de proposer une ventilation non invasive avant l’apparition d’une hypercapnie diurne et d’adapter les séances de kinésithérapie. Si vous remarquez que vous respirez nettement mieux assis que couché, ou que vous vous réveillez la nuit à bout de souffle, n’hésitez pas à le signaler explicitement : dans la maladie de Pompe, ce détail n’est jamais anodin.
Signes neurologiques et musculaires spécifiques : distinguer la dystrophie musculaire des ceintures
La maladie de Pompe tardive est souvent confondue avec une dystrophie musculaire des ceintures, car les deux pathologies partagent une faiblesse proximale, une marche dandinante et une élévation des CPK. Comment faire la différence ? En repérant certains signes neurologiques et musculaires plus spécifiques de la glycogénose de type II. L’atteinte des muscles respiratoires et paravertébraux est souvent plus marquée et plus précoce dans la maladie de Pompe que dans de nombreuses dystrophies.
Contrairement à certaines dystrophies musculaires, la maladie de Pompe ne s’accompagne pas d’atteinte cognitive ni de troubles sensoriels. Les nerfs périphériques sont globalement préservés, même si quelques études rapportent des atteintes neurogènes discrètes secondaires à la surcharge lysosomale. Le tonus musculaire est diminué (hypotonie) mais les réflexes peuvent être normaux ou légèrement diminués, sans signe pyramidal.
Sur l’IRM musculaire, le pattern d’atteinte est évocateur : muscles paraspinaux, diaphragme, psoas et muscles fessiers sont très tôt infiltrés de graisse, alors que certains groupes musculaires distaux restent relativements épargnés. L’électromyogramme (EMG) montre un tracé myopathique, parfois associé à des potentiels de fibrillation. Devant tout tableau de myopathie proximale inexpliquée, surtout s’il existe des signes respiratoires nocturnes ou une scoliose marquée, la maladie de Pompe doit être évoquée et un dosage enzymatique GAA demandé, même si une dystrophie des ceintures semble plausible.
Critères diagnostiques biologiques : dosage enzymatique GAA et test sur goutte de sang séché DBS
Le diagnostic de certitude de la maladie de Pompe repose sur la mise en évidence d’un déficit en alpha-glucosidase acide (GAA) et sur l’identification de mutations bialléliques du gène GAA. Ces examens spécialisés sont indispensables pour confirmer que les symptômes musculaires et respiratoires que vous présentez relèvent bien d’une glycogénose de type II et non d’une autre myopathie. Grâce aux progrès des méthodes de dépistage, le dosage enzymatique peut aujourd’hui être réalisé à partir d’une simple goutte de sang séché (DBS).
En pratique, le médecin commence souvent par des examens plus accessibles : dosage des créatine-kinases (CPK), transaminases, LDH, parfois électromyogramme. En cas d’anomalies évocatrices, il prescrit un dosage de l’activité GAA sur DBS, leucocytes ou fibroblastes cutanés. En parallèle, une analyse génétique moléculaire est lancée pour rechercher les mutations du gène GAA sur le chromosome 17q25.3. Associer enzymologie et génétique permet de fiabiliser le diagnostic et d’orienter le conseil génétique familial.
Activité résiduelle de l’alpha-glucosidase acide sur leucocytes et fibroblastes cutanés
Le dosage enzymatique de la GAA est l’étape clé. Il peut être réalisé sur différents types de cellules : leucocytes circulants, fibroblastes cultives à partir d’une biopsie cutanée, ou encore sur goutte de sang séché (DBS). Dans la forme infantile classique, l’activité enzymatique résiduelle est quasi nulle, généralement inférieure à 1 % de la normale. Dans les formes tardives, elle est réduite mais non abolie, souvent entre 3 et 30 % selon les études.
Le test sur goutte de sang séché a pris une place croissante car il est peu invasif, facile à transporter et compatible avec un dépistage néonatal élargi. Il permet une première orientation rapide : une activité GAA très basse sur DBS doit être confirmée sur leucocytes ou fibroblastes, afin d’exclure d’éventuels faux positifs liés à des variations techniques. À l’inverse, une activité normale rend très improbable une maladie de Pompe cliniquement significative.
Pour vous, cela signifie qu’un simple prélèvement sanguin peut désormais ouvrir la voie à un diagnostic précis, sans attendre des examens plus lourds. En cas de myopathie inexpliquée avec suspicion de glycogénose de type II, n’hésitez pas à demander si un test GAA sur DBS a été envisagé : c’est aujourd’hui l’un des outils les plus efficaces pour réduire l’errance diagnostique.
Élévation des créatine-kinases et transaminases : marqueurs de lyse musculaire chronique
Les créatine-kinases (CPK) sont des enzymes musculaires libérées dans le sang lors de lésions des fibres. Dans la maladie de Pompe, leur taux est généralement modérément à fortement augmenté, souvent entre 2 et 10 fois la normale, mais avec une grande variabilité individuelle. Cette élévation témoigne d’une lyse musculaire chronique, comparable à ce que l’on observe dans d’autres myopathies.
Les transaminases (ALAT, ASAT) et la LDH peuvent également être augmentées, ce qui peut orienter à tort vers une atteinte hépatique primaire. Il est donc essentiel d’interpréter ces marqueurs hépatiques en tenant compte du contexte musculaire : une élévation concomitante des CPK doit faire envisager une cause musculaire plutôt qu’une atteinte du foie lui-même. Dans la maladie de Pompe, l’hépatomégalie est liée à la surcharge glycogénique, mais la fonction hépatique reste souvent relativement préservée.
On peut comparer ces enzymes à des « signaux d’alarme » envoyés par les muscles dans le sang : plus la dégradation musculaire est importante, plus le signal est fort. Toutefois, un taux normal de CPK n’exclut pas formellement une maladie de Pompe, surtout dans les formes très modérées ou à évolution lente. C’est pourquoi ces marqueurs doivent être considérés comme des indices, et non comme des critères diagnostiques définitifs.
Analyse génétique moléculaire : mutations du gène GAA sur le chromosome 17q25.3
L’analyse génétique moléculaire du gène GAA est indispensable pour confirmer la maladie de Pompe et caractériser précisément le profil mutationnel. Plus de 400 variants pathogènes ou probablement pathogènes ont été décrits, incluant des mutations faux-sens, non-sens, des délétions ou des insertions. La maladie se transmet sur le mode autosomique récessif : vous devez donc hériter de deux copies altérées du gène (une de chaque parent) pour développer la maladie.
L’identification des mutations permet non seulement de confirmer le diagnostic, mais aussi de proposer un conseil génétique aux membres de la famille (dépistage des apparentés, diagnostic prénatal ou préimplantatoire si souhaité). Certains génotypes sont associés à une forme infantile très sévère, d’autres à des formes tardives plus modérées, mais la corrélation génotype-phénotype reste imparfaite. Des facteurs modificateurs et environnementaux interviennent également dans la variabilité des symptômes.
Sur le plan pratique, l’analyse génétique se fait à partir d’un simple prélèvement sanguin. Le laboratoire séquence l’ensemble du gène GAA, voire utilise des panels de gènes myopathiques ou un exome complet lorsqu’un diagnostic plus large est envisagé. Pour vous, disposer d’un résultat génétique clair permet de mieux comprendre l’origine de la maladie, d’anticiper le risque pour vos enfants et d’accéder plus facilement à certains essais cliniques ciblés.
Examens complémentaires paracliniques : IRM musculaire et électromyogramme myopathique
Au-delà des tests biologiques et génétiques, plusieurs examens paracliniques aident à préciser l’atteinte musculaire et à orienter vers une maladie de Pompe. L’IRM musculaire fournit une cartographie fine des muscles atteints, révélant des zones d’infiltration graisseuse et d’atrophie, tandis que l’électromyogramme (EMG) met en évidence un tracé myopathique typique. Ces outils ne posent pas à eux seuls le diagnostic, mais ils renforcent fortement la suspicion clinique et guident les prélèvements les plus pertinents.
Sur l’IRM, le pattern d’atteinte musculaire dans la glycogénose de type II est assez caractéristique : muscles paraspinaux, psoas, ilio-psoas, fessiers et muscles de la loge postérieure de la cuisse sont fréquemment touchés. À l’inverse, certains muscles distaux ou de la loge antérieure peuvent être relativement épargnés, surtout au début. Cette distribution sélective aide à distinguer la maladie de Pompe d’autres myopathies métaboliques ou dystrophiques, qui présentent des schémas d’atteinte différents.
L’électromyogramme montre un profil myopathique avec des potentiels d’unités motrices de faible amplitude et de durée courte, associés à un recrutement précoce. Des potentiels de fibrillation ou des ondes positives aiguës peuvent refléter une souffrance musculaire active. Bien que l’EMG ne soit pas spécifique de la maladie de Pompe, il confirme la nature myogène de l’atteinte et permet d’écarter une neuropathie périphérique ou une atteinte de la jonction neuromusculaire.
Dans certains cas, une biopsie musculaire peut encore être réalisée, en particulier lorsque les résultats enzymatiques ou génétiques sont équivoques. Elle montre alors des vacuoles lysosomales remplies de glycogène, visibles en coloration PAS (Periodic Acid-Schiff) et résistantes à la digestion par la diastase. Toutefois, avec l’essor des tests enzymatiques sur DBS et de la génétique moléculaire, la biopsie est de moins en moins nécessaire.
En combinant ces différents outils — clinique, biologie, imagerie, EMG et génétique — vous et votre équipe médicale pouvez poser plus rapidement un diagnostic de maladie de Pompe. C’est cette approche globale, intégrée, qui permet aujourd’hui de réduire l’errance diagnostique et d’initier plus tôt les traitements spécifiques, avec un impact concret sur la qualité de vie et le pronostic à long terme.